Learning how a BioMetAll calculation works¶
Objectives¶
In this first tutorial, the goal is to understand all the options that BioMetAll has to personalize a calculation.
The only mandatory input for a BioMetAll calculation is a structure of the biological system that will be object of study. It can be provided in a .pdb file or by indicating its 4-character PDB code. In the latter case, the structure will be downloaded from the Protein Data Bank and saved in a xxxx.pdb file on the working directory.
Tip
BioMetAll can handle files with several chains, but if you are interested specifically in one/several of them, removing the non useful chains in your .pdb file is recommended.
Along this use case tutorial, the chain A of the PDB code 1dhy (KKS102 BPHC enzyme) will be used.
1. Executing the program with default parameters¶
To execute the exploration of possible metal-binding zones, you simply should call the executable of BioMetAll followed by the .pdb file path or the 4-character PDB code:
biometall <path/to/the/input_file.pdb>
or
biometall 1dhy
You will obtain a list of solutions (i.e. the possible coordinating environments), followed by the coordinates of the center of the area, its radius and the number of probes which match the coordination criteria. In the case of 1dhy, 39 solutions are obtained, being the first one an area formed by 44 probes.
- The results of the execution will be shown on the screen and saved in a results_biometall_1dhy.txt file in the working directory.
- The different coordinating environments will be ordered by number of probes that match that environment.
- If one probe matches several coordinating environments (e.g. [HIS:145:A HIS:193:A HIS:194:A ASP:243:A] and [HIS:193:A HIS:194:A ASP:243:A]), that probe is accounted in all the environments it matches.
- The center and radius provided for each solution defines the minimum sphere which contains all the probes of that solution.
In addition to the .txt results file, you can obtain a .pdb file containing the different coordination areas with their corresponding centers. To do so, you should use the –pdb parameter:
biometall --pdb 1dhy
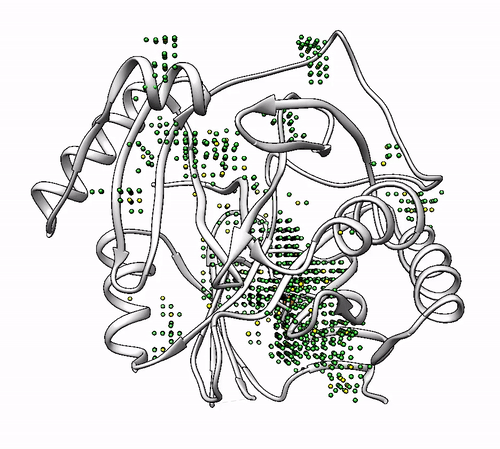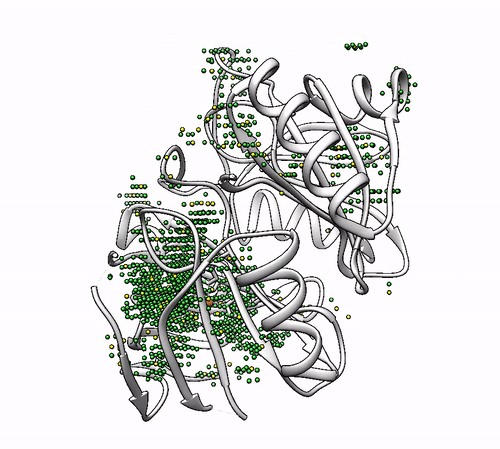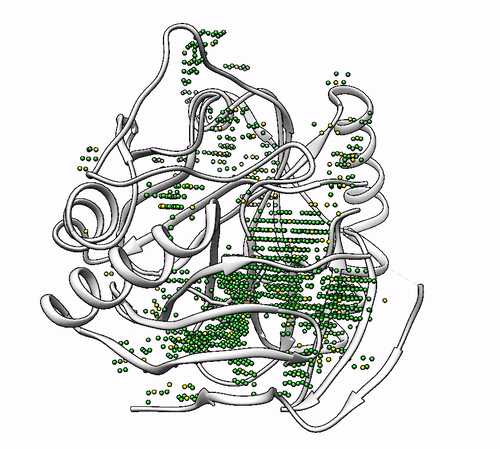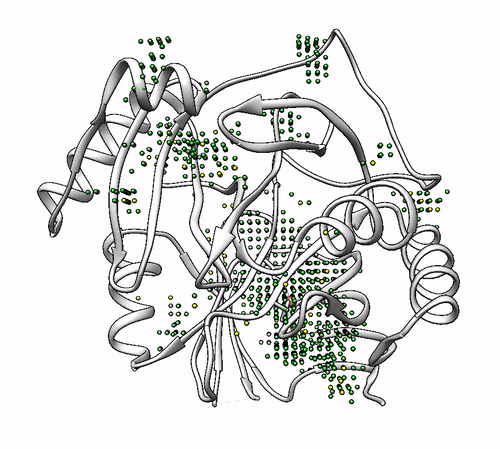
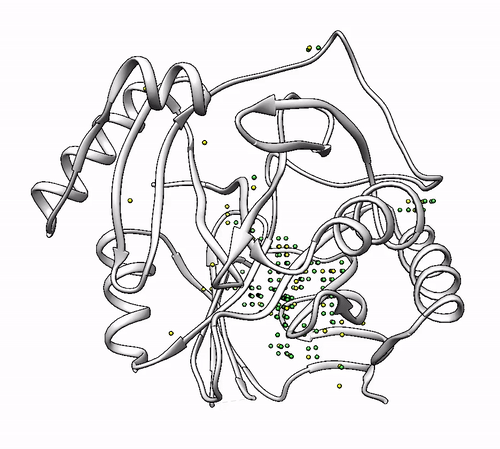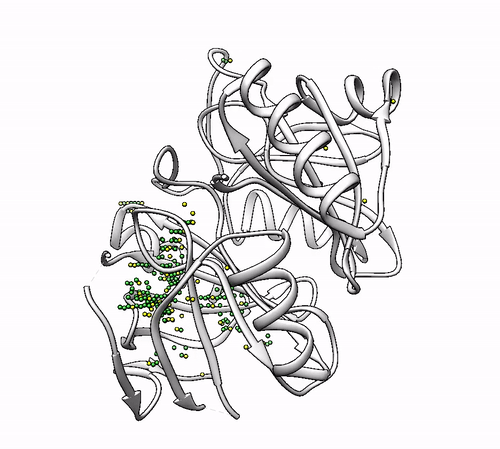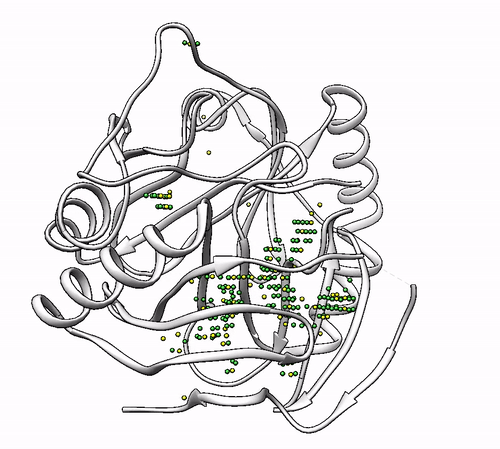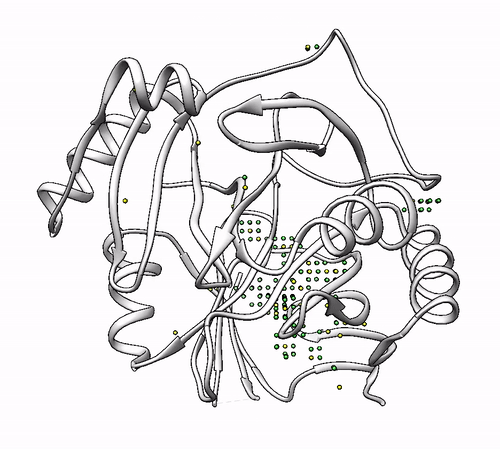
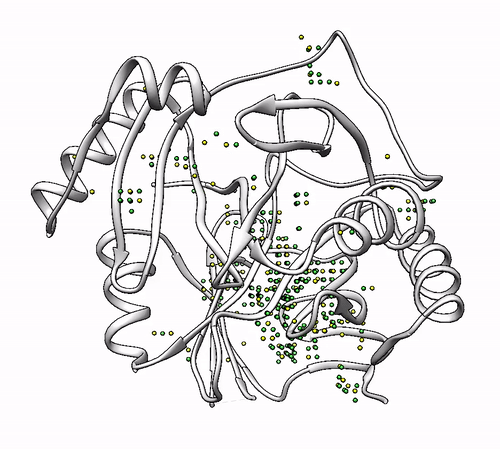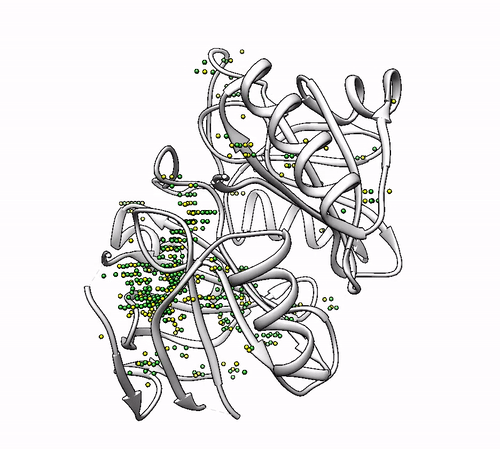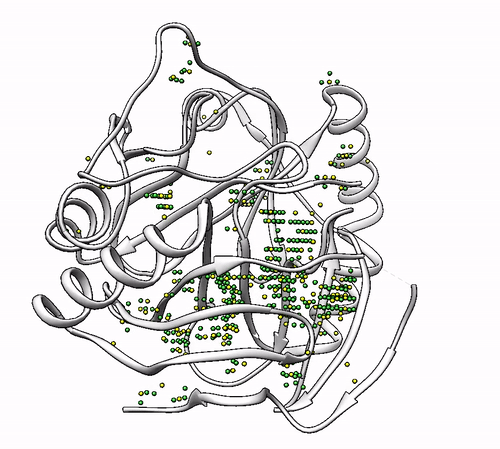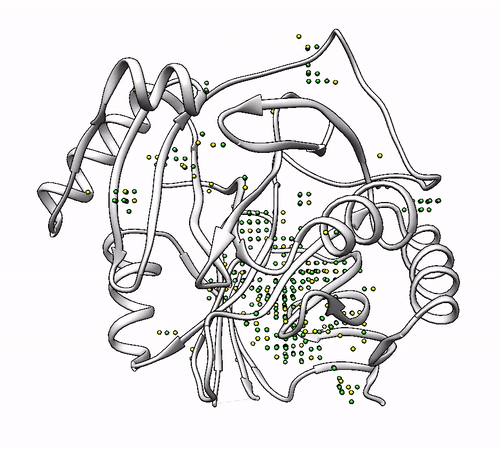
It will output a probes_1dhy.pdb that you can open together with your original .pdb file in any visualization program:
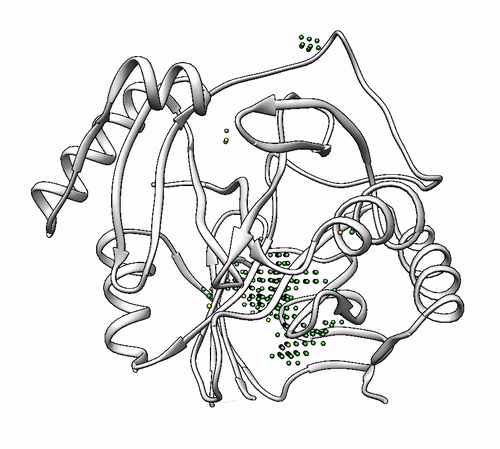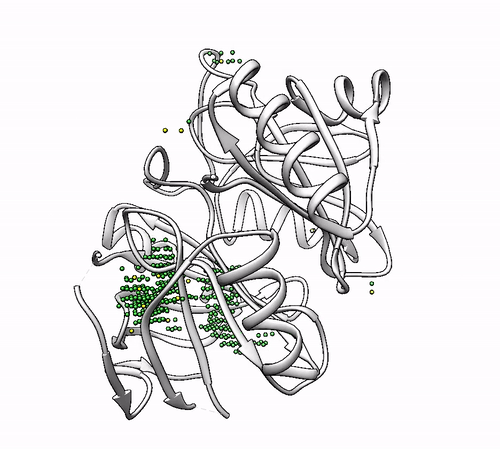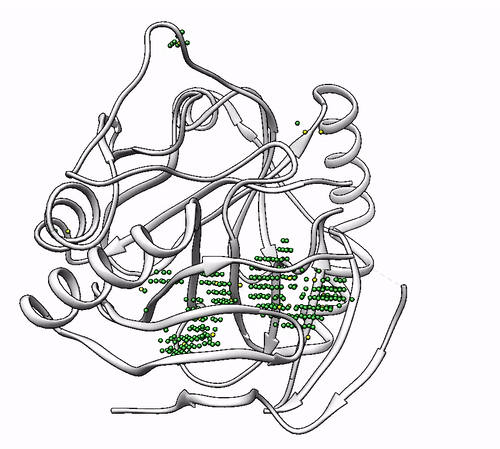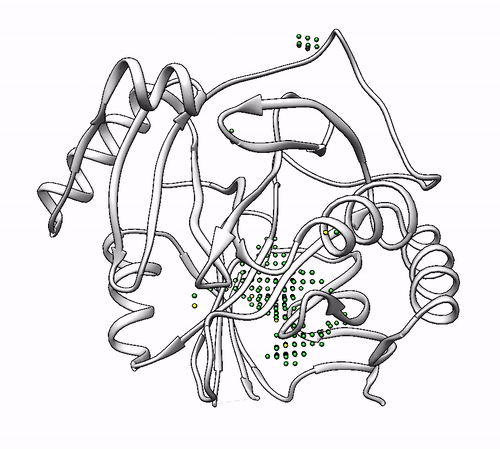
Every solution is stored as Xeon atoms (probes, depicted in green) and a Helium atom (center of each coordinating environment, depicted in yellow). Each solution is stored as a different residue in the probes_1dhy.pdb file, so you can easily see which probes corresponds to each one. For example, for the first coordinating environment (probes in green and center in yellow sphere):
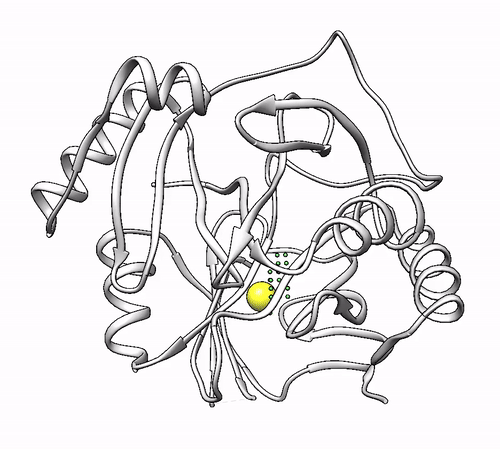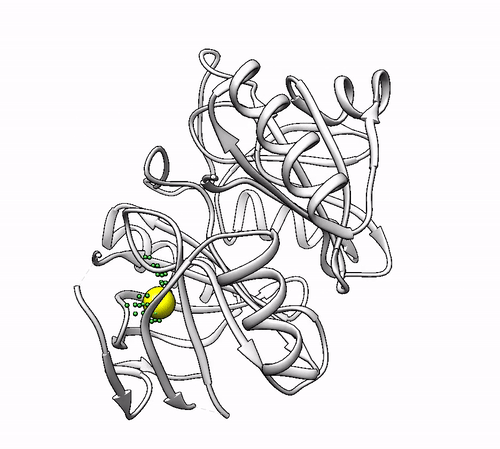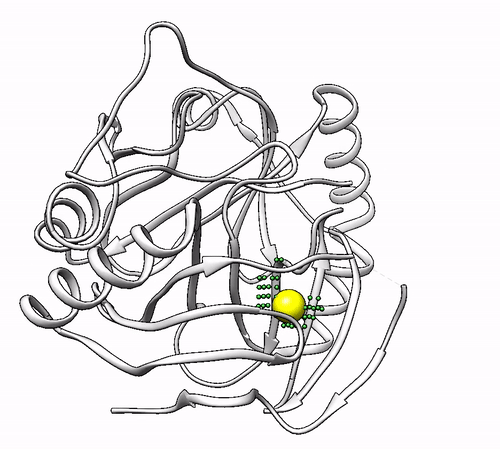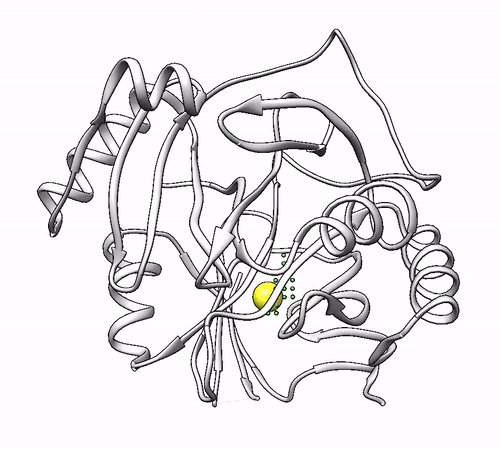
2. Executing with custom parameters¶
2.1. Filtering the results by number of probes (`–cutoff`)
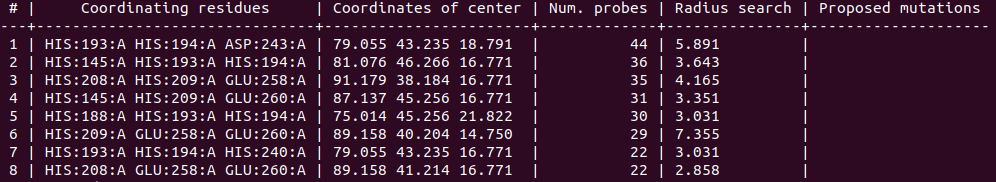
By default, the program proposes as solutions all the coordinating environments. If you want to obtain solutions only with a high number of probes (i.e. wide areas where the metal could coordinate), you should use the cutoff parameter. For example, you can use a cutoff of 0.5 to obtain solutions with a number of probes >= half of the probes of the solution with highest number. In the 1dhy example, only solutions with number of probes >= 22 will be provided:
biometall --cutoff 0.5 1dhy
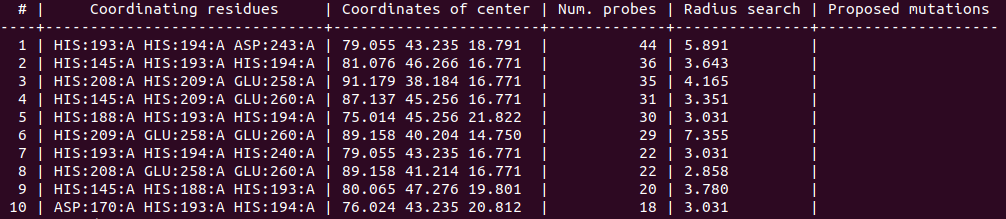
- To show solutions with a number of probes higher than 40% of the highest scored:
biometall --cutoff 0.4 idhy
2.2. Customizing which amino acids are considered as possible coordinators (`–residues`)
By default, the list of amino acids that are considered as potential metal coordinators is:
- ::
- ASP, HIS, GLU, CYS
You can modify this list using the –residues parameter. You should indicate the amino acids in their 3-letter code, enclosed between brackets [] and separated by commas (without spaces). For example, to search for coordinating environments formed by a (much) more extended set of amino acids, you could type:
biometall --pdb --residues [ASN,ASP,CYS,GLN,GLU,HIS,MET,SER,THR,TYR] 1dhy
2.3. Obtaining solutions with a minimum of coordinating amino acids (`–min_coordinators`)
By default, coordinating environments with a minimum of three amino acids are considered as valid solutions. If you want to change this number, it can be done with the –min_coordinators parameter.
- To show coordinating environments with a minimum of four amino acids:
biometall --pdb --min_coordinators 4 --residues [ASN,ASP,CYS,GLN,GLU,HIS,MET,SER,THR,TYR] 1dhy
2.4. Adding solutions that match backbone oxygen atoms (`–backbone`)
The usual case when searching metal-coordinating amino acids is to search for atoms of their side chains, and that is the default in BioMetAll calculations. However, in some cases could be useful to consider coordinations with backbone oxygens. To do so, you should use the –backbone parameter, followed by a list of the amino acids that you consider potential coordinators in their bakcbone. You should indicate the amino acids in their 3-letter code, enclosed between brackets [] and separated by commas (without spaces). Also, you can indicate the word ALL if you want to consider coordination with backbone oxygen of all amino acids.
For example, to consider coordinations with backbone oxygens of histidines besides the standard side chains coordinations, you should type:
biometall --backbone [HIS] 1dhy
To consider coordinations with backbone oxygens of all amino acids and a minimum of four coordinators:
biometall --min_coordinators 4 --backbone ALL --residues [ASN,ASP,CYS,GLN,GLU,HIS,MET,SER,THR,TYR] 1dhy
Warning
In this latter example, the minimum of coordinators refers to the sum of sidechains and backbone coordinations detected. An amino acid is counted double if it can be coordinated both by a sidechain atom and a backbone atom.
2.5. Defining a more dense (or scattered) grid (`–grid`)
BioMetAll defines a grid of points (i.e. probes) containing all the volume of your system. Each point of the grid is tested to see if it can be part of a coordinating environment. By default, points of the grid are generated with a separation of 1.0 Angstroms, but if you want a greater (or lower) detail, you can modify it using the –grid parameter. For example, to generate a grid with a separation of 0.5 Angstroms between probes:
biometall --grid 0.5 1dhy
Warning
grid parameter has direct implication in the computational time required to perform the calculation. It affects specially when –mutations option is used.
2.6. Defining a search zone (`–center` and `–radius`)
By default, the grid of probes is constructed to embed the whole system. If you have an specific zone that you want to search, it can be defined using the –center (for the center of coordinates of the zone) and –radius parameters. The coordinates should be enclosed between brackets [] and separated by commas (without spaces). For example:
biometall --center [84.98,42.82,16.04] --radius 10.0 1dhy
2.7. Defining how many processors are used for the calculation (`–cores`)
By default, BioMetAll calculations are run in a parallel mode using all the physical cores available in your computer. If for some reason (e.g. you are running BioMetAll in a cluster of computers) you want to change it, you can use the –cores parameters to define how many physical cores will be used. For example, to use two cores:
biometall --cores 2 1dhy